
Xeroderma Pigmentoso


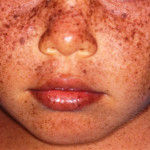


Genodermatose rara (1 para 250.000 nascimentos) de herança autossômica recessiva, marcada por exagerada fotossensibilidade, alterações pigmentares, envelhecimento precoce, neoplasias cutâneas e defeito na reparação do DNA por redução da atividade das endonucleases.
Etiopatogenia: O defeito básico está na incapacidade da reparação do DNA após exposição à radiação ultravioleta (UV). As células não conseguem excisar os dímeros pirimidínicos do DNA formados pela ação da luz UV. Quando em uma célula normal ocorre alterações estruturais no DNA induzidas pelo UV um sistema enzimático envolvendo enzimas tipo endonucleases, exonucleases, DNA polimerase e DNA ligase promovem a reparação de imediato. Entretanto no XP as células epidérmicas, os fibroblastos, os neurônios e os linfócitos são incapazes de promover o mecanismo de reparo do DNA por excisão, devido à diminuição da atividade destas enzimas. Acredita-se que estes dímeros sejam potencialmente carcinogênicos. Aproximadamente 80% dos pacientes com XP mostram um defeito no início da reparação por excisão do DNA. Na forma variante do XP (20% restantes) a reparação por excisão é normal. O defeito ocorreria mais tardiamente, na fase de reparo pós-duplicação do DNA.
Manifestações clínicas: As alterações cutâneas do xeroderma pigmentoso já estão presentes em 75% dos casos nos 3 primeiros anos de vida. Os pacientes apresentam queimaduras solares facilmente, fotossensibilidade aguda, sardas e xerose na face, dorso das mãos e antebraços. Observa-se progressivo envelhecimento cutâneo de forma prematura traduzido pelo afinamento da epiderme, elastose dérmica, lentigens solares, hipopigmentações e ceratoses actínicas. A fotofobia é marcante com conjuntivite, ceratite, opacificação da córnea e ectrópio. A malignidade cutânea é invariável e se deve às mutações UV induzidas. A idade média de aparecimento dos cânceres cutâneos é de 8 anos, ao contrário da população geral que é de 65 anos. Em ordem decrescente de frequência são reportados ceratoses actínicas, carcinomas basocelulares, carcinomas espinocelulares e melanomas.
Cerca de 5% dos pacientes desenvolvem melanomas. Os pais dos afetados, que são heterozigotos obrigatórios, são usualmente clinicamente normais, mas têm mostrado uma elevada incidência de câncer cutâneo. Envolvimento neurológico está presente em 20% dos pacientes. Manifesta-se por hiporreflexia, perda auditiva neurossensorial, alterações motoras, sensitivas e piramidais. Baseados no quadro clínico e no defeito molecular distinguem-se 7 subtipos ou grupos de complementação no XP, representados pelas letras A, B, C, D, E, F e G, além de uma forma variante, onde o defeito na reparação ocorre após a duplicação do DNA.
Muitos pacientes do grupo C (forma mais comum na Europa e USA) não mostram defeitos neurológicos. No grupo A doença neurológica se desenvolve antes dos 7 anos e no grupo D após esta idade.
A síndrome de De Sanctis-Cacchione corresponde a associação do XP com microcefalia, retardo no crescimento, deficiência mental, hipogonadismo e alterações neurológicas múltiplas. É a forma mais severa de XP. O diagnóstico pré-natal pela amniocentese é possível.
Tratamento: As lesões cutâneas são evitáveis desde que seja feita uma rigorosa prevenção contra a exposição aos raios solares, através de roupas, vidros das janelas da casa e dos carros, óculos e filtros solares adequados. Uma vez instalados, as ceratoses actínicas e os tumores cutâneos devem ser tratados com rigor desde o uso tópico do 5-fluoruracil até a excisão com a técnica de Mohs. Tem-se utilizados métodos como os peelings, a dermoabrasão e o uso de retinóides orais como o acitretin e a isotretinoina na tentativa de evitar o desenvolvimento de novos tumores cutâneos. A deterioração neurológica também pode ser reduzida pela proteção solar precoce.
